Introduction
The MDR (Medical Device Regulation) is currently a hot topic and is keeping manufacturers of medical products and their suppliers on their toes. There are many different reasons for this. On one hand, the timeline laid out by the EU is extremely ambitious and, on the other, it is not yet clear how much of what is detailed in the regulation will be implemented in practice.
What is the MDR?
The new European MDR replaces the existing MDD (Medical Device Directive) and AIMD (Active Implantable Medical Devices), which will now be combined in the MDR in future.
The advantage of the regulation is that the same aspects apply uniformly for all EU member states. In the two former directives, these had to be implemented in local law. This in turn led to differing assessments in the individual countries.
The goal of the MDR is to increase product safety for patients, which is at the heart of the whole regulation. Transparency should also be increased. A central database (EUDAMED) will be established, where everyone (including private individuals) can obtain information on every medical product approved in the EU.
Changes compared to the MDD
There are several changes in different areas. Four of the most important ones are detailed below.
Expanded scope of application
The AIMDs are now also regulated in the MDR. Moreover, products that do not necessarily have a medical use are also covered by the MDR. Examples here include cosmetic contact lenses, lasers for tattoo removal and implants for body modification.
Classification
The MDR now includes 22 rules for classifying medical products. In the MDD, there were only 18. This is one of the reasons why each medical product has to be newly assessed and classified.
Clinical evaluation of medical products
The requirements for the clinical data required for approval of a medical product are now more stringent. The MDR also describes in more detail which quality the data should have for the clinical assessment. The goal of this is to prove that the claimed effectiveness also exists in practice. Moreover, it should also be possible to exclude any unexpected or unacceptable side effects.
Market surveillance
Post market surveillance (PMS) is now more important than was previously the case. The data obtained in this way must also be incorporated in the clinical assessment of the products, which is then updated accordingly. It is also not sufficient to only monitor one’s own products. Comparable products from competitors also have to be included in PMS. If incidents occur, consideration must be given as to whether measures also have to be taken for own products.
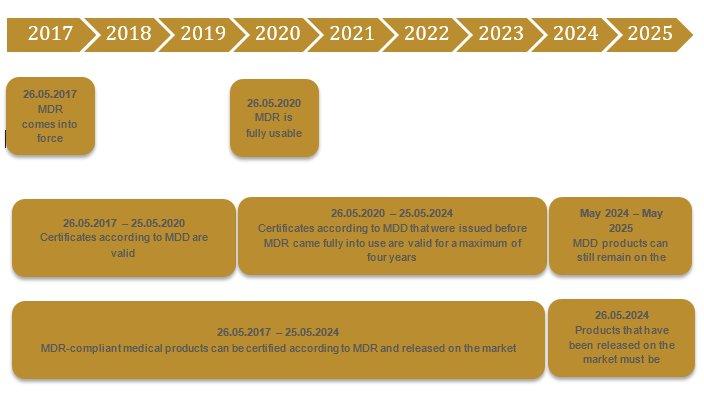
The transitional period from the MDD to MDR is three years, up to 26.05.2020. In theory, it has already been possible to certify products according to the MDR for some time. However, there is still only a limited choice of notified bodies that are certified according to the MDR. To our knowledge, a total of 34 companies have applied for certification as a notified body. Two have been certified up to now. Taking into account the timeline and the host of medical product manufacturers who require new certificates, the transitional period appears to be overly ambitious. The EU is aware of this. In the past, there have also been rumors that the MDR will be postponed by three years.
How is artimelt dealing with the MDR
With this in mind, artimelt has also kept up to date with the MDR and its consequences via various seminars and through service providers. We are thus well aware of the importance of the MDR for our customers in the medical branch and are prepared to support them as much as we can and share what we know about the MDR.
artimelt has created its own database for products and raw materials in order to maintain an organized and easy to manage overview of the situation. This database should cover the different requirements of the various regulations, whether REACH, MDR, California Proposition 65 or individual queries such as the absence of heavy metals or other critical substances.
Furthermore, artimelt is also committed to making available as much data on biocompatibility as is necessary. This simplifies the creation of technical documentation in diverse product classes and is a good starting point for creating a clinical assessment of your medical product.
We are keeping our eye on the ball here, and are looking forward to sharing information on the MDR and how we can master these challenges together.
artimelt AG
Wassermatte 1
CH6210 Sursee
Telefon: +41 (41) 92605-00
Telefax: +41 (41) 92605-29
http://artimelt.com
Chief Revenue Officer
Telefon: +41 (41) 92605-00
Fax: +41 (41) 92605-29
E-Mail: christoph.lang@artimelt.com